The process was, as every year, a lengthy one, and the core of our project. Our lab team was thankfully able to enter the lab this summer and carry out a great deal of work to validate our dry lab designs, which will be detailed below. We are pleased to report that despite not having enough time to achieve our end goals, we believe our project was at the very least partially successful.
After pinpointing the problem of Carbapenem-resistance Enterobacteriaceae (CRE), we investigated several leads we had on detection methods. Ideas such as using bait-prey interactions between biosensors proved far too complicated to design and using specifically engineered bacteriophages to infect CRE with genes encoding fluorescent reporters would have proven too lengthy and expensive. Instead, our attention was drawn to an area previously explored by both teams and academics from our university – the use of CRISPR as a detection tool by engineering a guide RNA (gRNA) able to sense the presence of our target gene - known as CRISPR activation, or CRISPRa for short. As for the latter, as mentioned on the Description page, we settled upon the detection of a particular Carbapenemase known as OXA-48 (Fig.1). This was because its activity is slower than other Carbapenemases, which made it more difficult to quickly detect using available tests such as the Carba NP test – a result requires sampling and sequencing for a positive result.
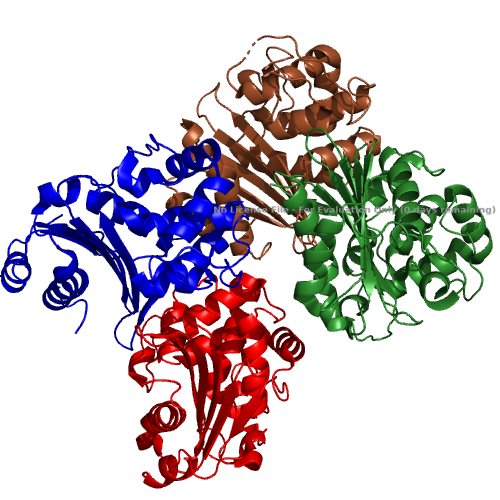
Fig. 1. The overall structure of the OXA-48 Carbapenemase. It is made up of 4 identical chains (each labelled in a different colour) which assemble to form the full protein.
Detecting DNA, however, has a chance of giving false positives as DNA can be left over from dead bacteria after, for example, treatment of a surface with biocides, resulting in unnecessary measures being taken. We therefore endeavoured to detect the mRNA of OXA-48, due to its relatively short lifespan being indicative of recently alive CPE within the sample (protein sequence obtained from [1]).
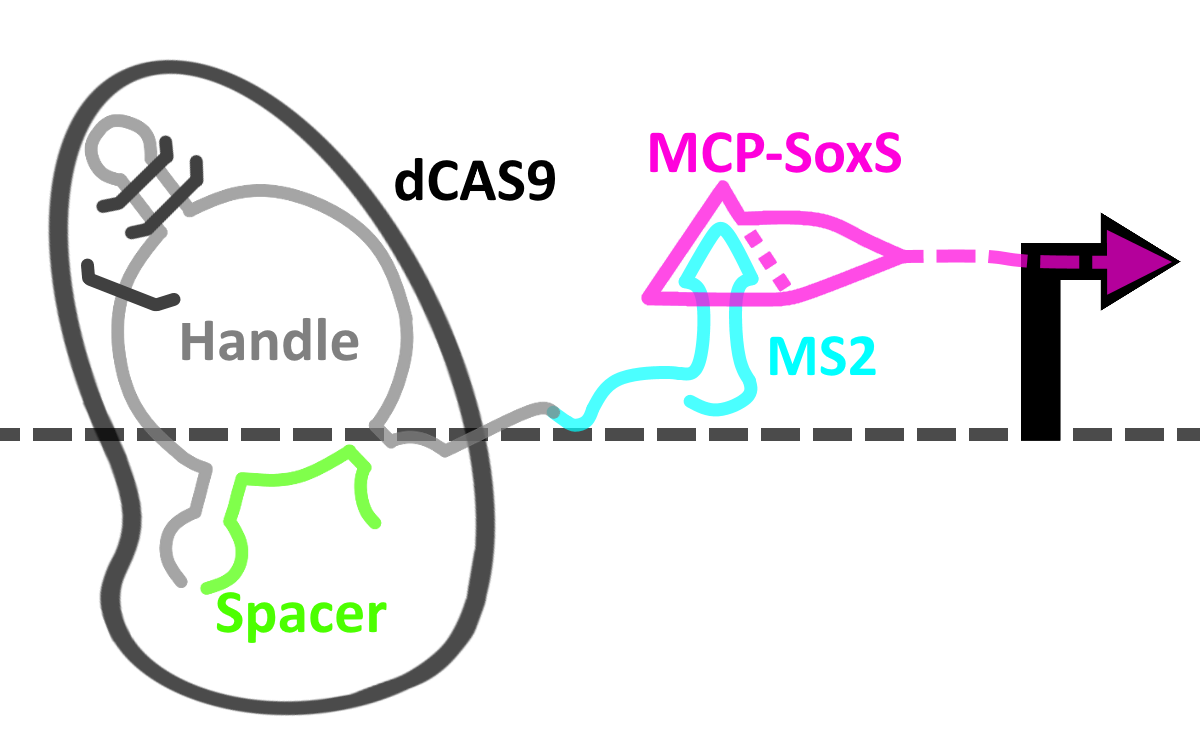
Fig. 2. Plasmid map of our ‘operator’ plasmid, encoding our cgRNAs and the CRISPRa system.
Our CRISPRa system was engineered with all the controls in mind – each component of the circuit is thus inducible. To make this easier, we settled upon using E. coli cells of the MG1655-Z1 line, as they contain the elements TetR, LacI and AraC, eliminating the need to introduce them ourselves (obtained from [2]). Instead of using a normal Cas9 protein, however, we opted to use a deactivated variant (dCas9), which has had its nuclease function abolished – the protein would simply stay bound to the DNA sequence indicated in the gRNA.
The gRNA we engineered to sense the presence of the OXA-48 mRNA can be extended to create a binding domain for the RNA-binding protein MS2 without compromising its ability to bind to dCas9. MS2 would provide a scaffold for the binding of a fusion protein composed of MCP and the transcriptional enhancer SoxS [3] – effectively causing the upregulation of transcription downstream of the dCas9 binding site (protospacer adjacent motif or PAM). By placing the PAM just upstream of the promoter of our reporter, we can cause its greatly increased production.
In a CRISPR system, a gRNA recognises its PAM sequence via Watson-Crick base pairing in order to guide the Cas9 protein to its cleavage site. We added another condition to this binding – the presence of our target mRNA – this turned our gRNA into a conditional gRNA (cgRNA).
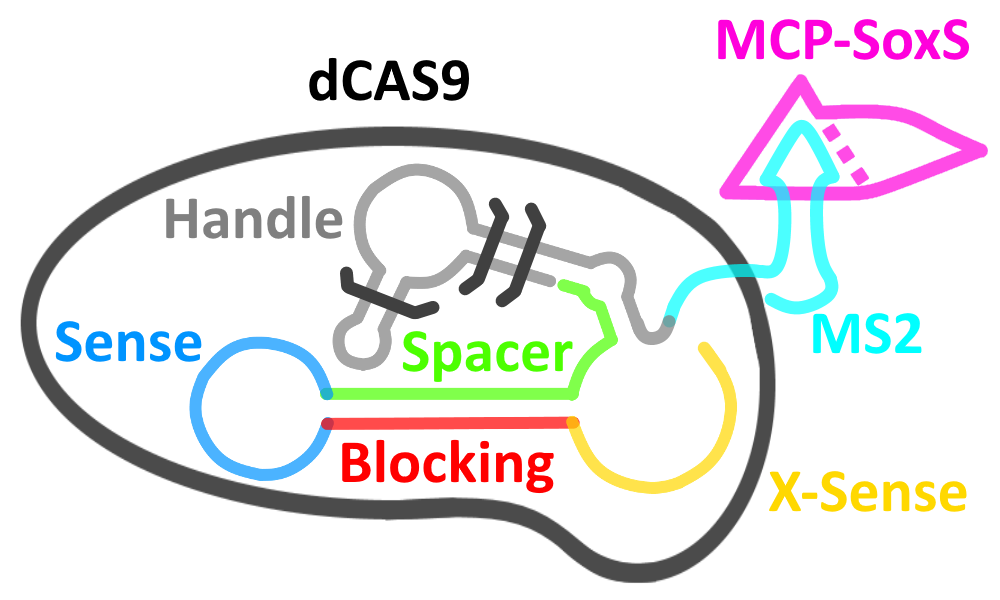
Fig.3. Representation of our switch in an ‘off’ state.
As seen in the above figure, the cgRNA is made up of a number of distinct domains. In its natively folded state, the dCas9 binding handle (grey) adopts a conformation which prevents its binding to dCas9 – the system is thus in an ‘off’ state.
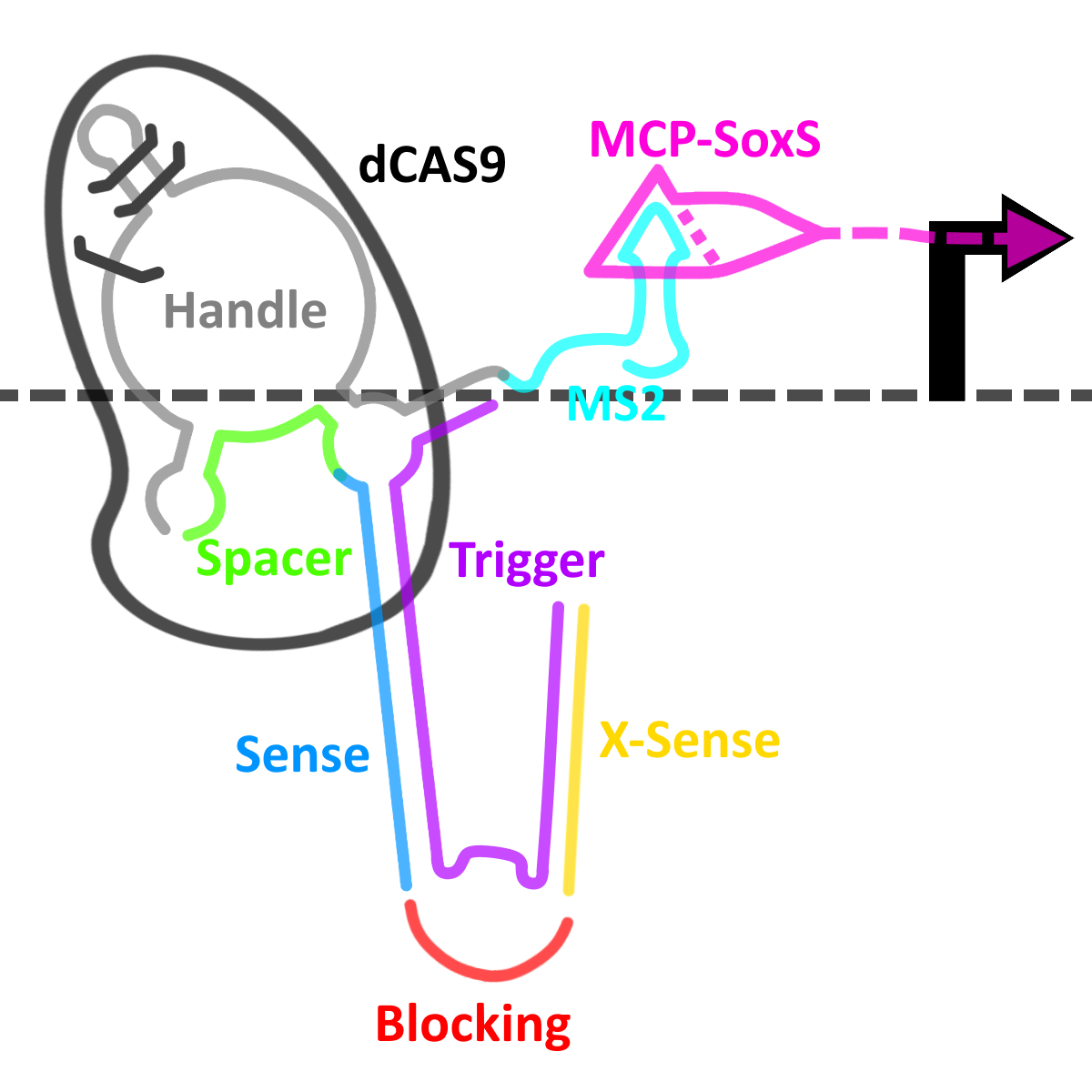
Fig. 4. Representation of our switch in an ‘on’ state.
However, when the OXA-48 mRNA is present (the trigger, in purple), the two RNA molecules anneal, causing a significant conformational change in the secondary structure of the cgRNA, allowing it to bind dCas9 and switching the system into its ‘on’ state, allowing the complex to bind to the PAM and activate transcription as detalied in the CRISPRa section above.
We simulated the secondary structure of our cgRNA in its natively folded form and when annealed to the mRNA in NUPACK, as seen in Fig. 5.
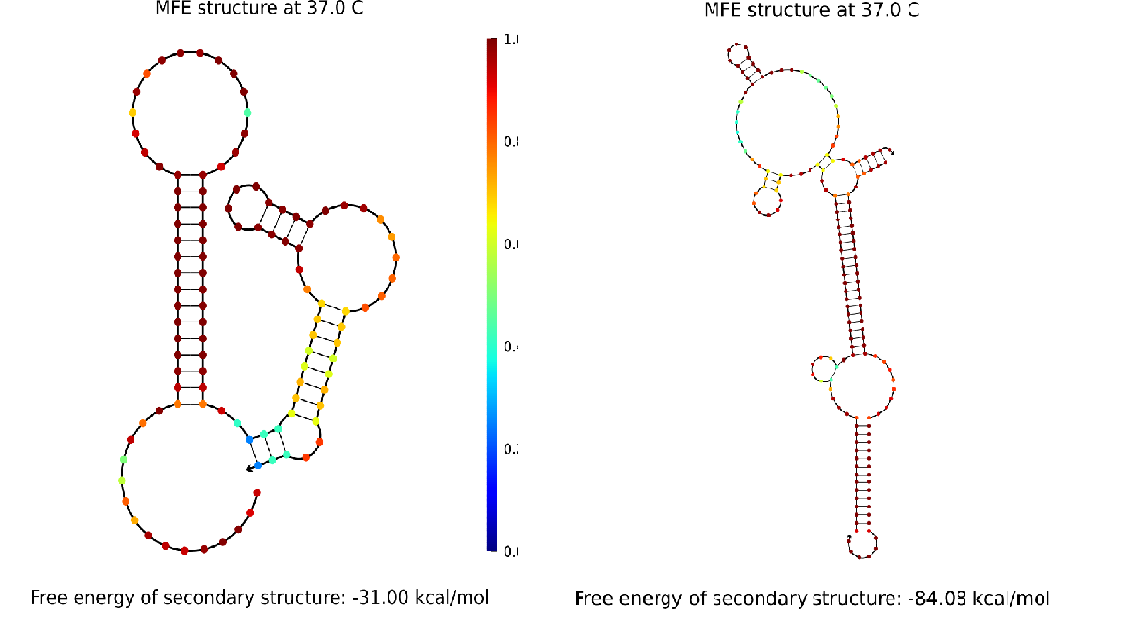
Fig. 5. (left) Diagram of the secondary structure of the cgRNA in its natively folded state. (right) Diagram of the secondary structure of the cgRNA when annealed to the target mRNA.
On the left of the figure, the dCas9 binding handle is misfolded, but the bonds in the blocking domain, shown in blue, are not particularly stable – this is indicative of the fact that the cgRNA can be brought out of this conformational state. On the right of the figure, the conformation is drastically changed, and the dCas9 binding handle (at the top in this figure) has folded correctly.
All of these components and more were cloned into one of two plasmids, as seen in the schematic in Fig.6. As for reporters, we chose to keep the original sfGFP as well as introducing a new one – a fluorescent RNA aptamer known as iSpinach; this RNA produces a strong green fluorescent signal when combined with the non-toxic fluorogenic compound DFHBI.
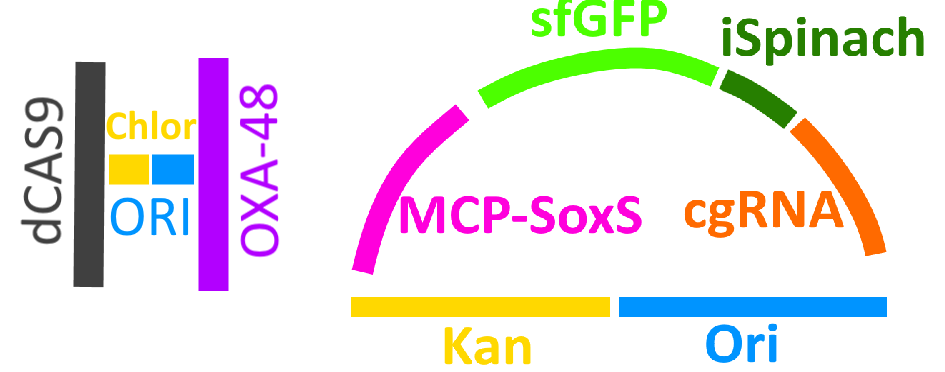
Fig. 6. Schematic diagram of the two plasmids in our system.
Above is the schematic of the helper plasmid, encoding our trigger as well as dCas9 and a chloramphenicol resistance gene to assist with selection. Below is the schematic of a reporter plasmid, containing the two reporters – sfGFP and iSpinach – as well as a cgRNA, the MCP-SoxS CRISPRa system, and a kanamycin gene to assist with selection.
The design is modular to allow for a large amount of control (Fig. 7)– the production of the cgRNA is inducible by arabinose (1) and the production of the trigger is inducible with anhydrotetracycline (ATC, 2), while both are required for the activation of dCas9 (3). Only when this occurs does MCP-SoxS (4) exercise its enhancing activity upon the sfGFP promoter (5).
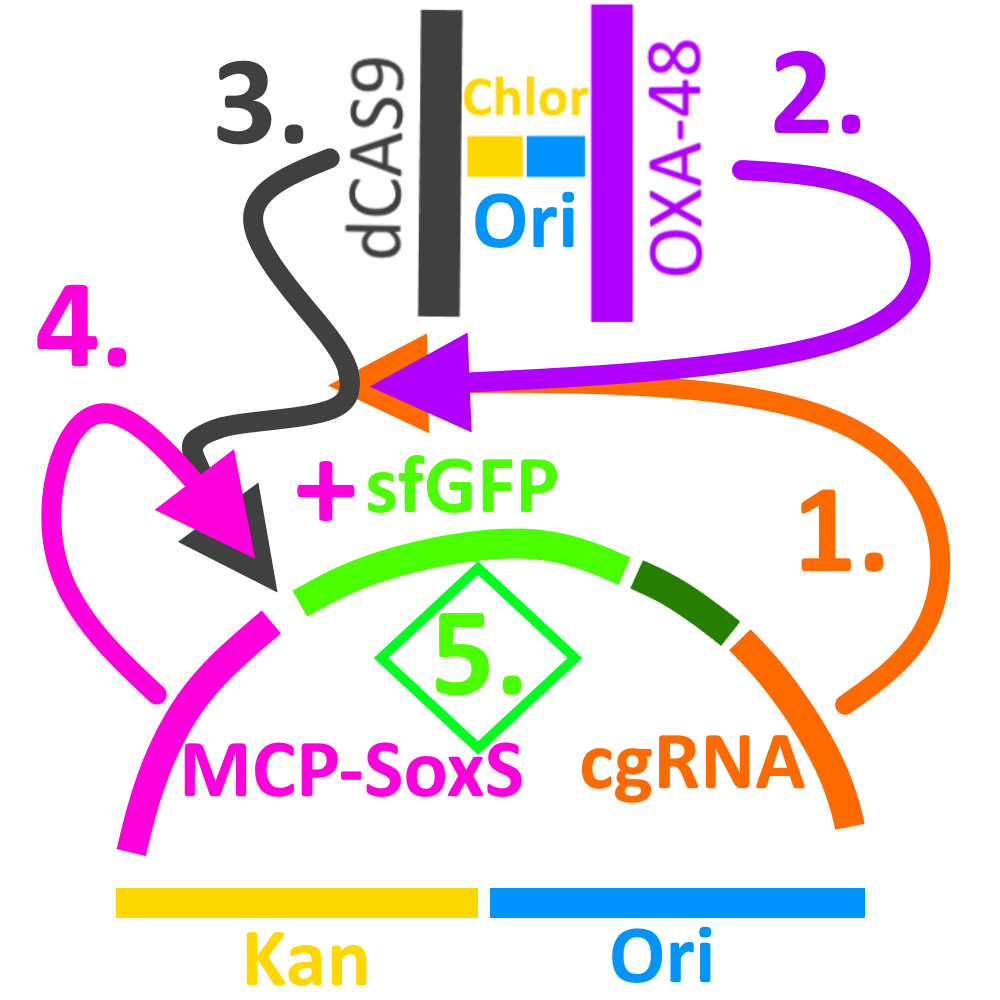
Fig. 7. Schematic representation of the cgCRISPR sensing device.
The flow of our experiments can be seen in the figure below - the lab work was split into four stages: foundation, improvement, engineering, and production. Unfortunately, due to time constraints and unforeseen circumstances brought by supply chain issues, we were only able to assemble the first reporter plasmids in time, reaching the first iteration cycle.
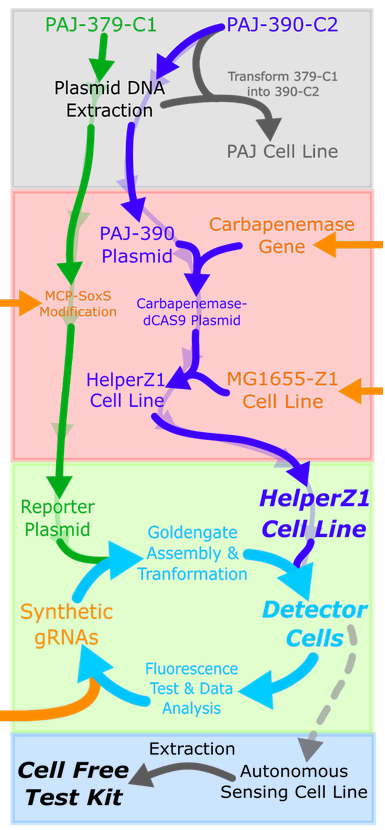
Fig. 8. Representation of our lab plan. Top to bottom: foundation phase, improvement phase, engineering phase, production phase.
We began by extracting the necessary components from existing plasmids (PAJ-379-C1 and PAJ-390-C2 [4]) from Professor Alfonso Jaramillo’s lab at the University of Warwick using PCR – this gave us the linearised backbones which we would use in assembling the plasmids in the above sections. Our inserts were provided by IDT and included the OXA-48 gene (sequence obtained from [1]). It lacked a ribosome binding site as we did not want to proliferate Carbapenemase resistance during our project. Other parts included the MCP-SoxS gene (sequence obtained from [3]). We used Gibson and Goldengate assembly techniques to assemble our helper and reporter plasmids, respectively.
Afterwards, MG1655-Z1 cells were transformed with the helper plasmid, giving rise to the helper cell line. This cell line would be transformed with different versions of the reporter plasmid containing different cgRNAs to screen for the one which offered the greatest difference in fluorescence between a positive and a negative result.
As alluded to in the diagram, as well as on our Human Practices page, our system was designed to work in an in vitro scenario; the iSpinach aptamer has been designed to function optimally in vitro. While we were not able to pursue this cell-free application due to time and materiel constraints, we are confident that a future iGEM team could validate the theoretical basis behind this device working cell-free.
A detailed rundown and timeline of our lab work can be found on our Experiments page.
In order to find out which of our designed cgRNAs was most appropriate despite a lack of data, we prepared a mathematical model using which we tested a promising sequence against a number of cgRNAs with mismatching spacers. The results of the first comparison can be observed in Fig.9 below – while there is a significant level of activation, the longer sensing loop could cause unintended activation, which is why we followed this up by testing a sequence with a shorter sensing loop which would give more specificity to the target sequence.
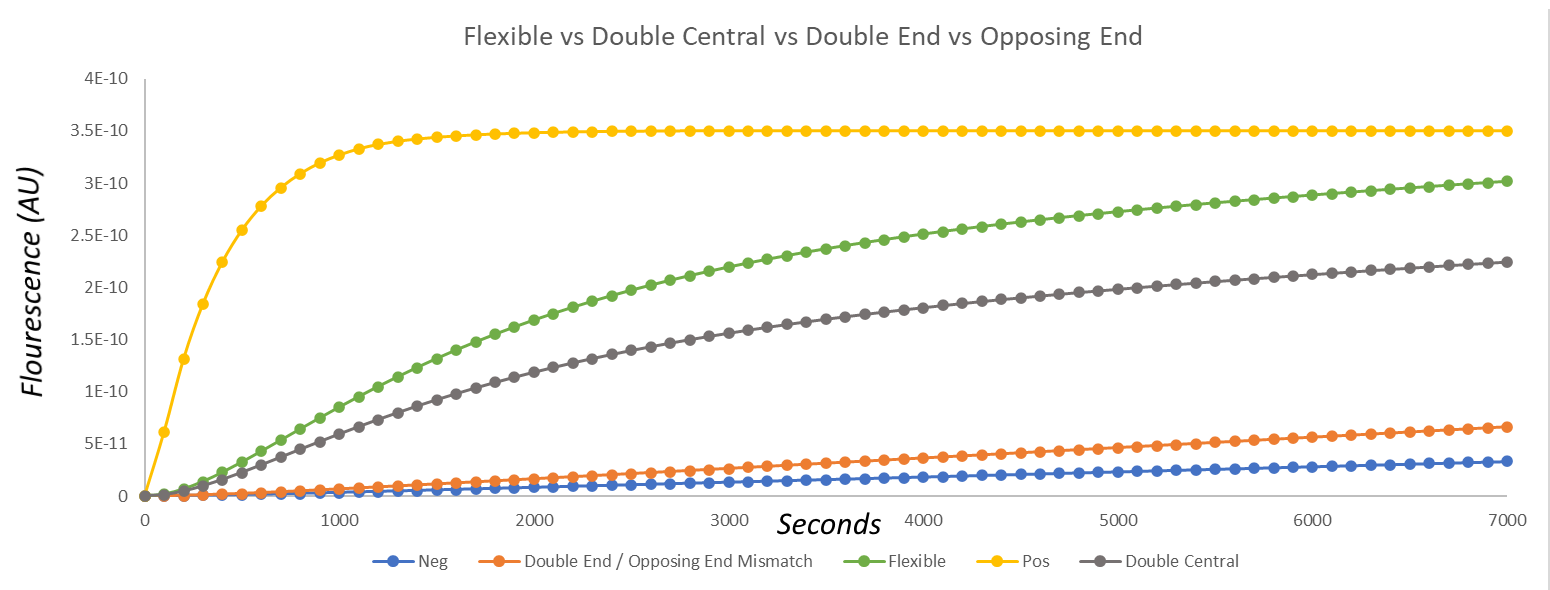
Fig.9. Graph of the results of the modelling of fluorescence over time when using a cgRNA with a longer sensing loop (green), when compared against mismatching cgRNAs (orange, grey) and a negative (blue) and positive (yellow) control.
The comparison of the other cgRNA to a mismatch using our model (Fig.10) yielded better results, however. There is a large difference in fluorescence between the use of the cgRNA we tested when compared against both the negative control and against a mismatching gRNA. Moreover, over time the fluorescence appears to approach the value exhibited by the positive control! This was, to us, indicative of the fact that the increased specificity had no negative effect on activation.
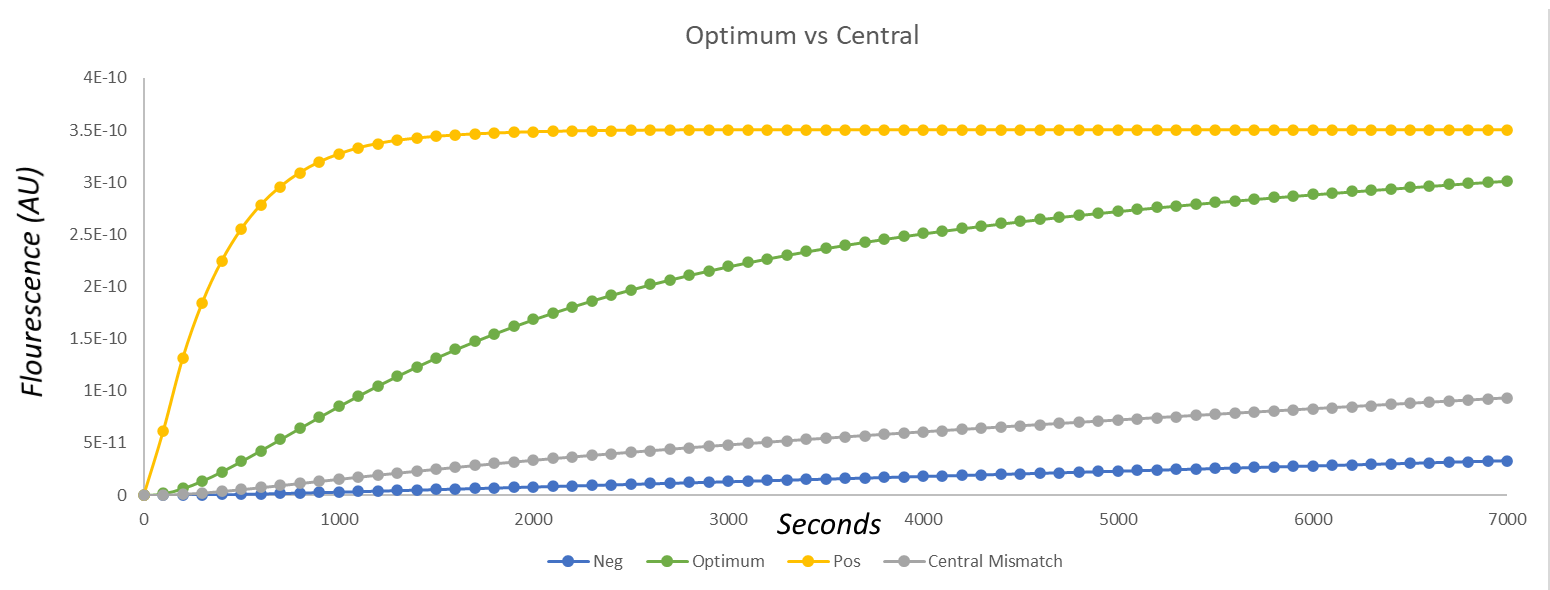
Fig.10. Graph of the results of the modelling of fluorescence over time when using a cgRNA with a shorter sensing loop (green), when compared against a mismatching cgRNA (grey) and a negative (blue) and positive (yellow) control.
We decided to investigate the sensitivity of this particular cgRNA as well and simulated its activity with different ratios of cgRNA:trigger as evidenced in Fig. 11. The results of this were satisfactory, as when compared to the absence of the trigger, the 1:1 ratio yielded a large increase in observed fluorescence.
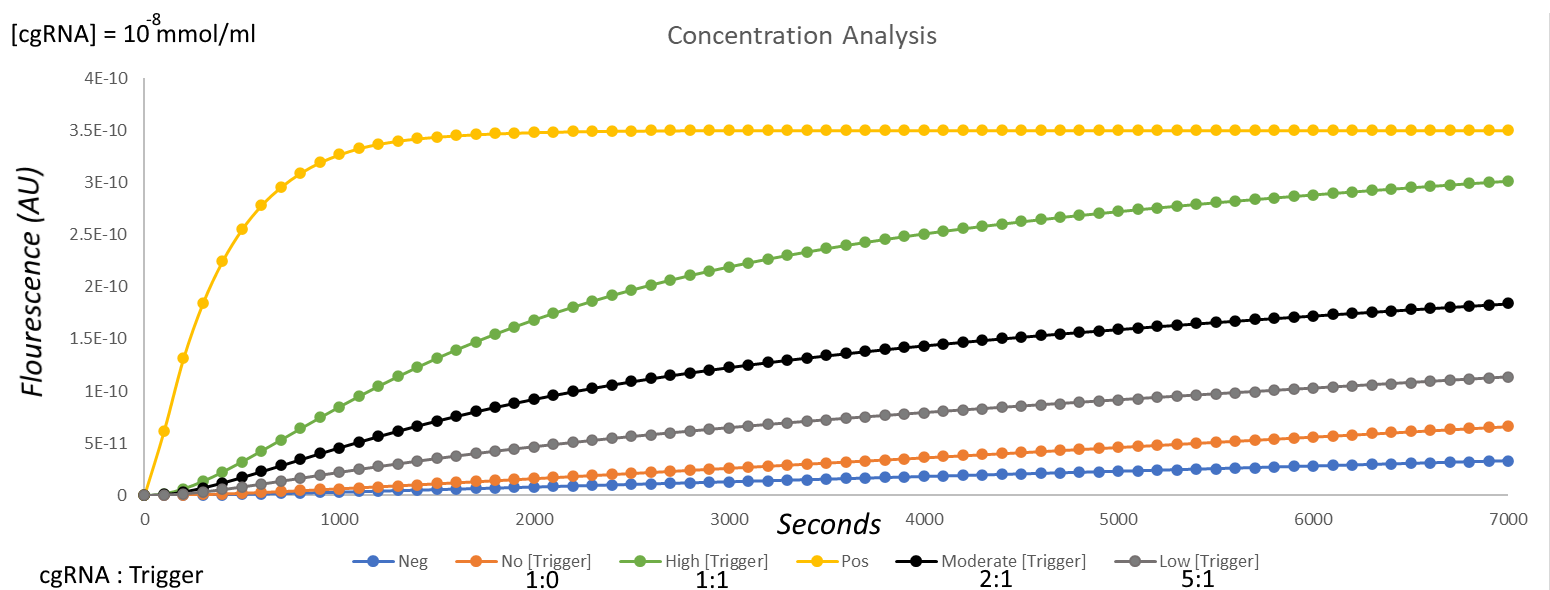
Fig 11. Comparison of fluorescence using the cgRNA with the shorter sensing loop at different concentrations – no trigger (orange), 1:1 cgRNA:trigger (green), 2:1 cgRNA:trigger (black), 5:1 cgRNA:trigger (grey).
As expected though, as the ratio of cgRNA to trigger increased, less activation was observed – this raises an issue when working with low concentrations of OXA-48 mRNA in the sample; nonetheless, we believe that this gRNA would make a fine candidate for further development.
See the reactions and values for our model below:
[1] Mohammadi Bandari, N. Keyvani, H. Abootaleb, M. Klebsiella pneumoniae strain TFH21 OXA family beta-lactamase (blaOXA) gene, partial cds. Available at: https://www.ncbi.nlm.nih.gov/nucleotide/MZ275246.1 [Accessed 19/10/2021]
[2] Sekar, K. Gentile, AM. Bostick, JW. Tyo, KE. N-Terminal-Based Targeted, Inducible Protein Degradation in Escherichia coli. PLoS One. 2016 Feb 22; 11(2):e0149746. doi: 10.1371/journal.pone.0149746. eCollection 2016. PONE-D-15-42990 [pii] PubMed 26900850. Available at: https://www.addgene.org/65915/ [Accessed 20/10/2021]
[3] Dong, C. Fontana, J. Patel, A. et al. Synthetic CRISPR-Cas gene activators for transcriptional reprogramming in bacteria. Nat Commun 9, 2489 (2018). https://doi.org/10.1038/s41467-018-04901-6
[4] Gallzi, R., Duncan, J. N., Rostain, W., Quinn, C. M., Storch, M., Kushwaha, M., Jaramillo, A., Engineered RNA-interacting CRISPR Guide RNAs for Genetic Sensing and Diagnostics, The CRISPR Journal (2020), 3 (5), pp. 398-408, doi: http://doi.org/10.1089/crispr.2020.0029